When precedent becomes problematic
We often encounter scientists already using qPCR assays in their research that rely on assays they’ve pulled from the literature. Why switch to something new when there are already established assays in the field? We’ve mentioned before how difficult it can be to measure “reality” when using microbial profiling methods (see blog posts on metagenomics and 16S amplicons and our preprint). Measuring reality is crucial as it can prevent the development of new diagnostics or pervert our understanding of how the microbiome impacts health. Here we’re sharing a cautionary example illustrating how legacy assays could be warping reality and how we address those issues in our advanced qPCR assays.
Fusobacterium nucleatum (Fn) has a long history of being around when things are going bad – increased abundance associated with tumors, potential tumorigenic activity, presence in polymicrobial infections in the mouth. However, despite a long history and growing consensus, individual studies have reported a wide range of associations between Fn and disease states. At Branchoint, we think this variation and inconsistency may simply reflect outdated assays and illustrates two of key principles that give our technology and approach an advantage. I’m going to tackle them both here: (1) microbial taxonomy, and (2) legacy qPCR assays. Buckle up for a deep dive!

Accurate Microbial taxonomy.
Ironically, many of the bacteria we have studied the most have names that make the least sense. They are often historical artifacts that do not reflect current biology. In the case of Fn, when originally discovered the only way to identify a bacterium was through a microscope or on a petri dish based on growth traits like those described in Figure 1. Differences in those traits led to classification of Fn into 4 subspecies. When 16S sequencing was finally employed they all had (essentially) identical 16S sequences further cementing that these trait differences were part of the larger definition of Fusobacterium nucleatum.
In the literature all 4 subspecies – F. nucleatum subsp. nucleatum, F. nucleatum subsp. animalis, F. nucleatum subsp. vincentii, and F. nucleatum subsp. polymorphum – were commonly summarized as Fusobacterium nucleatum. You can visualize that in this modified figure where all of the subspecies are summarized as a single 16S rRNA gene sequence entry at the top of the tree.
Thankfully, we have come a long way since identifying bacteria in this manner. At Branchpoint, we have embraced the use of genome taxonomy – a taxonomic system based on whole genome similarity and multilocus, conserved gene phylogeny – hosted by the Genome Taxonomy Database (GTDB) to differentiate species of bacteria. Using this approach, we can clearly distinguish the 4 subspecies and accurately place additional species into a rigorously defined (but flexible) context.

GTDB not only split the Genus Fusobacterium into multiple genera to reflect the diversity between type species but also redefined the subspecies as standalone species with distinct names to reflect their genetic difference – F. nucleatum, F. animalis, F. polymorphum, and F. vincentii. We can also place those species into context relative to each other – like the presence of F. animalis on a distinct branch from the others and include several uncultivated lineages (e.g., F. nucleatum_E) that fall between the named isolates. Lumping together these distinct genetic lineages into a single entity may obscure meaningful signals that could explain why results are inconsistent across studies and prevent a deeper understanding of how these lineages impact health.
These historical challenges underscore the necessity for Branchpoint’s genomic taxonomy approach. Where once there was uncertainty, we now can apply modern genomic databases to deliver up-to-date assays that reflect taxonomic reality.
Legacy qPCR Assays.
Scientists can be a stubborn bunch. If you have used an assay for years with an established precedent in the literature, it is going to take a good reason to change things up. But relying on legacy can lead you astray. To illustrate how this can happen and why it is important to fix, I did a deep(er) dive on those assays to highlight where they come from and how their use may play into the uncertainty of results in the past and present. If this was already a deep dive, let’s touch the bottom! Let’s follow the paper trail that brought us to the current state of Fn qPCR assays.
A recent study by Eisele et al. (2021) in Clinical Colorectal Cancer used qPCR primers for quantification in a cohort of 105 treatment-naïve CRC patients. The manuscript cited one research paper as the source of the qPCR assay sequences: Tunsjø et al. (2019)
Tunsjø et al. (2019) sourced their primer sequences from Flanagan et al. (2014), who in turn sourced their sequences from Casterllarin et al. (2012).Casterllarin et al. (2012) performed an early metagenomic and metatranscriptomic analysis of frozen tumor and healthy colon tissue samples to look for differences in the microbiome. They compared their short 75bp RNA reads to NCBI RefSeq and saw the most matches to F. nucleatum subsp. nucleatum (ATCC 25586). And then to get quantitative data designed qPCR assays – here is the quote from the manuscript:
To design the qPCR primers and probe, we gathered the 51,677 read-pairs from tumor sample 1 that matched F. nucleatum and performed a local de novo assembly using SSAKE (Warren et al. 2007) to obtain 861 total contigs, ranging in length from 100 to 1433 bp. The majority of these contigs matched genes encoding F. nucleatum ribosomal RNAs and proteins, but we also obtained 82 contigs that gave BLASTN (Basic Local Alignment Search Tool) alignments of 80% or greater sequence identity to other F. nucleatum protein-coding genes. A 161-bp contig that returned a high-quality BLAST match (95% identity) to the nusG gene (GenBank accession AAL94126.1) of F. nucleatum and no match to any gene of any other species, was used as the target for designing a qPCR (Taqman, ABI) primer/probe set.
Sequences
Fusobacteria forward primer, 5′ CAACCATTACTTTA ACTCTACCATGTTCA 3′
Fusobacteria reverse primer, 5′ GTTGACTTTACAGAAGGAGATTATGTAAAA ATC 3′
Fusobacteria FAM probe, 5′ TCAGCAACTT GTCCTTCTTGATCTTTAAATGAACC 3′
Casterllarin et al. (2012)
A first generation meta-omics analysis, mapping a 161-bp fragment to a reference genome, that had no in silico cross-reactivity to NCBI in pre-2012, is the basis for qPCR assays used until at least 2021. Designed using the best data available at the time, the assays from Casterllarin et al. held up remarkably well. But we’ve come a long way in the last decade, and it’s time to revamp with the benefit of modern genomic databases. These primers have actually been used to quantify the Fusobacterium spp. as they can amplify the other subspecies in pure culture.
Tunsjø et al. without much explanation in 2019 drop the FAM probe component and make a modification to the reverse primer:
F. nucleatum-F, 5’ CAACCATTACTTTAACTCTACCATGTTCA 3’
F. nucleatum-R. 5’ TACTGAGGGAGATTATGTAAAAATC 3’
Tunsjø et al. (2019)
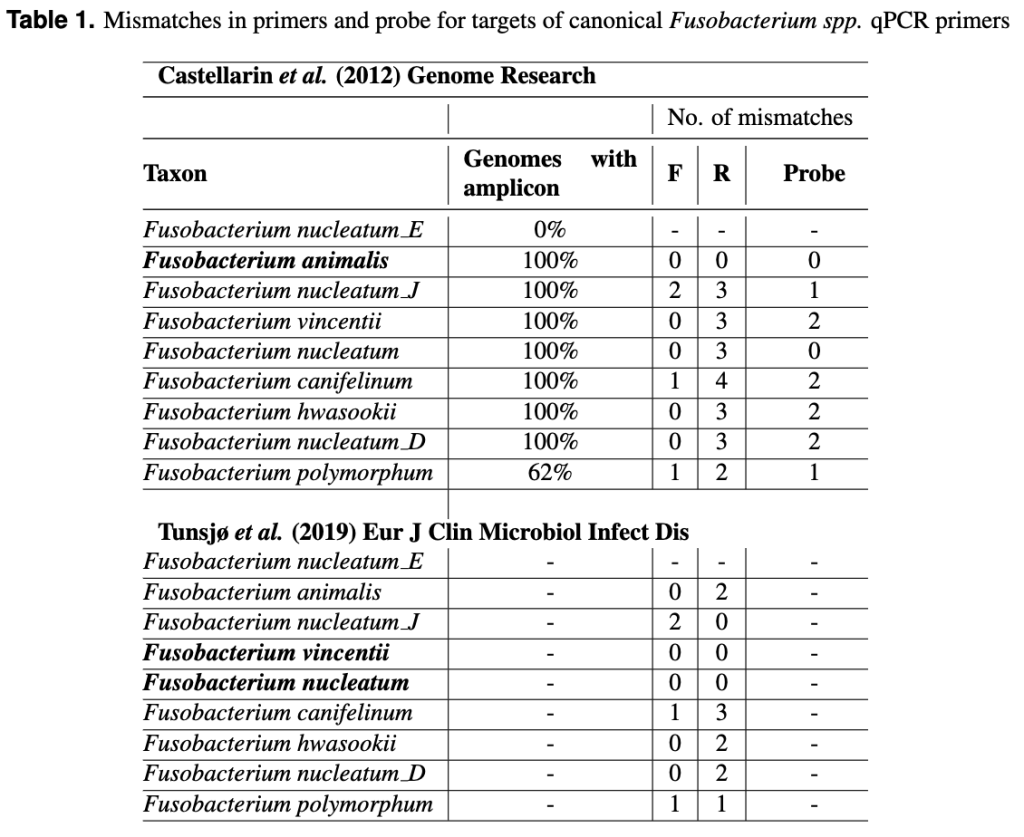
So why do all these details matter? It’s because these legacy primer and probes sequences have an impact on the research of today. The reverse primer from Casterllarin et al. (2012) has 3-4 mismatches in most of the Fusobacterium species and 1-2 mismatches in the probe sequence, but has exact matches for F. animalis. These mismatches represent a genetic discrepancy that can lead to inaccurate measurements of bacterial abundance as primers prefer identical sequences over those with mismatches. Working with primers and probes with that many mismatches is never linear. In this example, a perfect match with F. animalis would have a greater amplification efficiently compared to other members of the genus, while the probe would likely miss F. vincentii altogether due to mismatches.
This might explain the changes made by Tunsjø et al. (2019) to the reverse primer which should fare better by more effectively detecting F. nucleatum and equally amplifying F. vincentii. However, other species would still contribute an inconsistent signal and the lack of probe may reduce specificity and prevents multiplexing.
Especially when contemplating a bacterium that may cause tumors, research that has applied sequencing-based profiling have always returned to quantification using qPCR assays. So why might these small changes make a big difference? They could explain results like this from Eisele et al. (2021):
“Across Fn abundance groups, no statistically significant differences were observed for clinicopathologic or demographic characteristics. Although a higher proportion of Fn negative/low cases were diagnosed with tumors of the colon (71.1%) compared with Fn high cases (50.0%), these findings only reached marginal statistical significance (P = .06).”
Eisele et al. (2021)
Or discussion points like: “The primer set used in our study specifically quantifies Fn and is unable to detect Fusobacteria species on a larger taxonomic level.” Which for the quoted study and primer set is incorrect.
Branchpoint Solution.
This is what we specialize in at Branchpoint. Teasing apart these historical confusions and making qPCR assays that are up-to-date and exact, providing world-class quantification you can count on. We have validated qPCR assays with fluorogenic probes for F. animalis, F. canifelinum, F. nucleatum, F. nucleatum_E, F. nucleatum_J, F. polymorphum, and F. vincentii, with more on the way. And they are available to help you measure reality in your microbiome research! To learn how Branchpoint’s assays can bring precision to your microbiome research, contact us today.